Overview
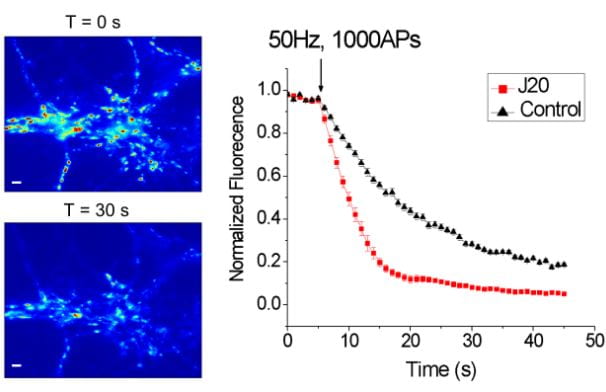
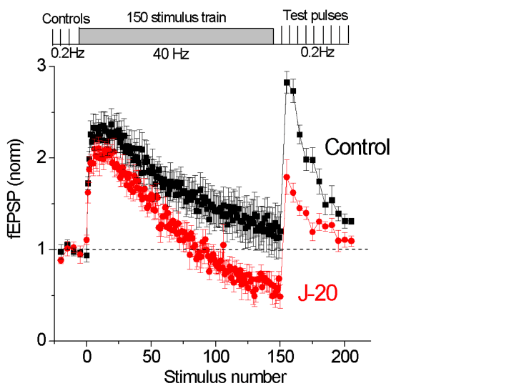
Synaptic dysfunction is one of the hallmarks of Alzheimer’s disease (AD) pathology, leading to deficits of memory and cognition. In particular, defects in synaptic vesicle recycling have been suggested to represent one of the earliest synaptic defects in AD models. Supported by funds from a New Investigator Award from the Alzheimer’s Association, we studied synaptic dysregulation in an APP transgenic mouse model of AD revealing a marked increase in a particular form of rapid synaptic depression at glutamatergic synapses. This form of synaptic depression is associated with fewer synaptic vesicles available at presynaptic terminals and we recently found that the kinetics of vesicle recycling is abnormal in this model of AD.
Thus, we hypothesize that synaptic depression in AD is a result of disrupted vesicle recycling. A significant loss of available vesicles due to recycling defects at glutamatergic synapses could explain at least some of the cognitive deficits suffered by patients with AD. This would represent a significant advancement in our understanding of the disease’s progression. Moreover, because defects in synaptic vesicle dynamics and recycling may be one of the earliest pathologies in AD, these processes could serve as targets for crucial early intervention and novel therapeutic strategies.
Progress
The main goal going forward is to define what leads to vesicle recycling defects in AD models, and how these disruptions may lead to AD pathophysiology. This project will utilize analyses of single vesicle recycling and release in two different rodent models of AD, with subsequent studies of implications the abnormal vesicle recycling has at neuronal and circuit levels. We have also begun to investigate the role of glia in abnormal synaptic transmission in AD models.